Geral
Publicado em 17/05/2025, às 18h54 Aryela Souza
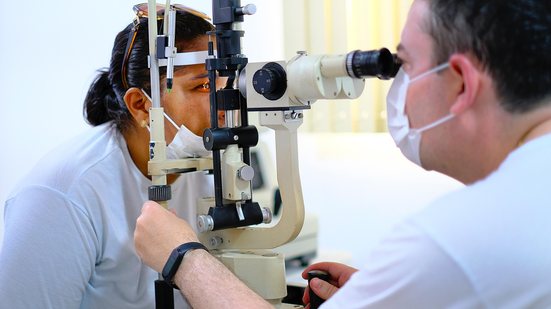
Cientistas da Universidade Federal do Rio Grande do Norte (UFRN), em parceria com a Universidade Johns Hopkins, nos Estados Unidos, identificaram duas novas mutações genéticas associadas à síndrome de Alport. A condição hereditária pode afetar rins, audição e visão, muitas vezes sem sintomas evidentes nos estágios iniciais.
O estudo, publicado na revista científica BMC Genomics, foi realizado com famílias dos municípios de São Miguel e Santo Antônio do Salto da Onça, no interior do estado. A pesquisa utilizou técnicas avançadas de sequenciamento genético e trouxe novas perspectivas sobre o desenvolvimento da síndrome em populações isoladas, além de contribuir para diagnósticos mais precoces e ações preventivas.
O que é a síndrome de Alport
Descrita pela primeira vez em 1927, a síndrome é causada por mutações nos genes COL4A3, COL4A4 e COL4A5, responsáveis pela produção do colágeno tipo IV — uma proteína essencial à membrana basal glomerular, estrutura dos rins que filtra impurezas do sangue. Quando essa proteína é produzida de forma incorreta, pode ocorrer perda progressiva da função renal. A condição também pode comprometer a audição e a visão, geralmente de forma silenciosa.
As novas mutações descobertas
A equipe do Instituto de Medicina Tropical da UFRN identificou duas mutações inéditas:
Uma no gene COL4A3, que interrompe precocemente a leitura do DNA e impede a produção adequada da proteína;
Outra no COL4A4, que modifica a estrutura do gene e resulta em uma versão incompleta da proteína.
As alterações foram encontradas em famílias com histórico de casamentos entre parentes próximos, o que aumenta o risco de doenças genéticas recessivas em comunidades isoladas.
Como foi feito o diagnóstico
Os cientistas utilizaram o sequenciamento completo do exoma — técnica que permite analisar todas as regiões do DNA relacionadas à produção de proteínas — e a metodologia runs of homozygosity (ROH), que detecta segmentos herdados idênticos do pai e da mãe.
A pesquisa mostrou que os quadros mais graves surgem quando o paciente herda duas cópias da mutação (uma de cada genitor), mas pessoas com apenas uma cópia alterada também podem desenvolver sintomas, em geral mais leves ou com início tardio.
Próximos passos
Os pesquisadores continuam acompanhando as famílias participantes e pretendem expandir o estudo para outras comunidades com características semelhantes. A ideia é identificar portadores das mutações antes do surgimento dos sintomas, o que permitirá um acompanhamento clínico mais eficaz e melhores perspectivas de tratamento.
O trabalho integra a tese de doutorado de Washington Candeia de Araújo, sob orientação da professora Selma Jerônimo, diretora do IMT/UFRN. Também contribuíram o oftalmologista Carlos Alexandre Garcia e a médica Raquel Araújo Costa Uchoa.
Classificação Indicativa: Livre






